An autoradiograph is an image on an X-ray film or nuclear emulsion produced by the pattern of decay emissions from a distribution of a radioactive substance. Alternatively, the autoradiograph is also available as a digital image, due to the recent development of scintillation gas detectors or rare-earth phosphorimaging systems. The film or emulsion is apposed to the labeled tissue section to obtain the autoradiograph. The auto- prefix indicates that the radioactive substance is within the sample, as distinguished from the case of historadiography or microradiography, in which the sample is marked using an external source. Some autoradiographs can be examined microscopically for localization of silver grains in which the process is termed micro-autoradiography. For example, micro-autoradiography was used to examine whether atrazine was being metabolized by the hornwort plant or by epiphytic microorganisms in the biofilm layer surrounding the plant.
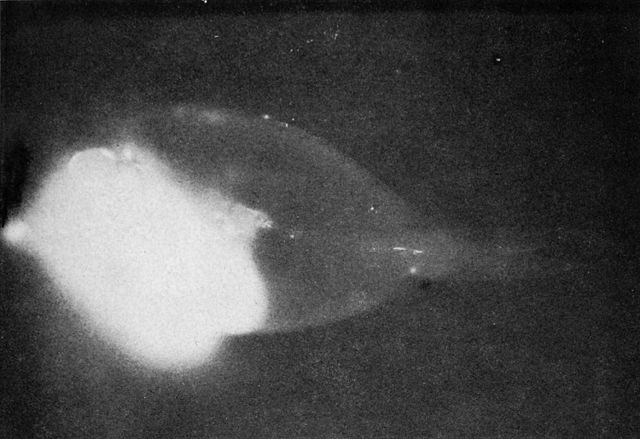
A radioactive surgeonfish makes its own X-ray. The bright area is a meal of fresh algae. The rest of the body has absorbed and distributed enough plutonium to make the scales radioactive. The fish was alive and apparently healthy when captured.
A nuclear emulsion plate is a type of particle detector first used in nuclear and particle physics experiments in the early decades of the 20th century. It is a modified form of photographic plate that can be used to record and investigate fast charged particles like alpha-particles, nucleons, leptons or mesons. After exposing and developing the emulsion, single particle tracks can be observed and measured using a microscope.
Schematic edge-view cross section of nuclear emulsion, not to scale.
Nuclear Emulsion Stack
Physicist Kinoshita Suekiti at the University of Manchester in 1910
Marietta Blau




