Platelet
Videos
Photos
Platelets or thrombocytes are a component of blood whose function is to react to bleeding from blood vessel injury by clumping, thereby initiating a blood clot. Platelets have no cell nucleus; they are fragments of cytoplasm derived from the megakaryocytes of the bone marrow or lung, which then enter the circulation. Platelets are found only in mammals, whereas in other vertebrates, thrombocytes circulate as intact mononuclear cells.

Image from a light microscope (500 ×) from a Giemsa-stained peripheral blood smear showing platelets (small purple dots) surrounded by red blood cells (large gray circular structures)

Platelets derive from multipotent marrow stem cells.
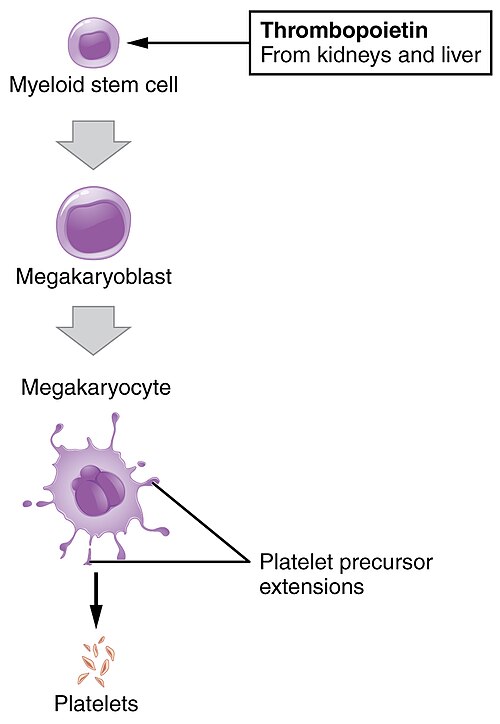
Platelets extruded from megakaryocytes

Scanning electron micrograph of blood cells. From left to right: human erythrocyte, activated platelet, leukocyte.
Blood
Videos
Photos
Blood is a body fluid in the circulatory system of humans and other vertebrates that delivers necessary substances such as nutrients and oxygen to the cells, and transports metabolic waste products away from those same cells.

Venous (darker) and arterial (brighter) blood

Two tubes of EDTA-anticoagulated blood. Left tube: after standing, the RBCs have settled at the bottom of the tube. Right tube: Freshly drawn blood
A scanning electron microscope (SEM) image of a normal red blood cell (left), a platelet (middle), and a white blood cell (right)

Vertebrate red blood cell types, measurements in micrometers