Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.

Part of a radioactively labelled sequencing gel
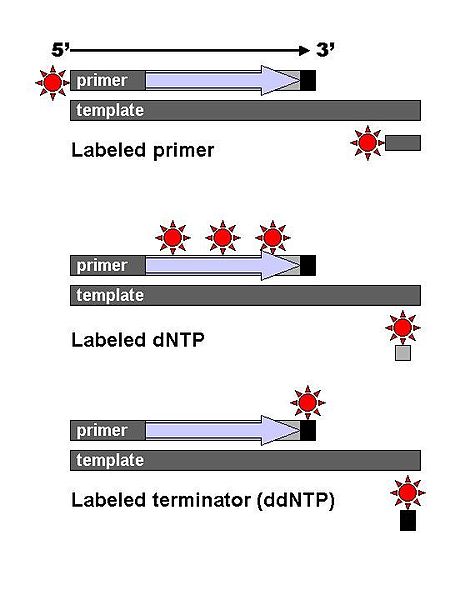
DNA fragments are labelled with a radioactive or fluorescent tag on the primer (1), in the new DNA strand with a labeled dNTP, or with a labeled ddNTP.
Sequence ladder by radioactive sequencing compared to fluorescent peaks
Capillary electrophoresis
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
An example of the results of automated chain-termination DNA sequencing.
Frederick Sanger, a pioneer of sequencing. Sanger is one of the few scientists who was awarded two Nobel prizes, one for the sequencing of proteins, and the other for the sequencing of DNA.
History of sequencing technology
Genomic DNA is fragmented into random pieces and cloned as a bacterial library. DNA from individual bacterial clones is sequenced and the sequence is assembled by using overlapping DNA regions.