Human genome
Videos
Page
The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.

TSC SNP distribution along the long arm of chromosome 22 (from https://web.archive.org/web/20130903043223/http://snp.cshl.org/ ). Each column represents a 1 Mb interval; the approximate cytogenetic position is given on the x-axis. Clear peaks and troughs of SNP density can be seen, possibly reflecting different rates of mutation, recombination and selection.

Populations with a high level of parental-relatedness result in a larger number of homozygous gene knockouts as compared to outbred populations.

A pedigree displaying a first-cousin mating (carriers both carrying heterozygous knockouts mating as marked by double line) leading to offspring possessing a homozygous gene knockout
DNA
Videos
Page
Deoxyribonucleic acid is a polymer composed of two polynucleotide chains that coil around each other to form a double helix. The polymer carries genetic instructions for the development, functioning, growth and reproduction of all known organisms and many viruses. DNA and ribonucleic acid (RNA) are nucleic acids. Alongside proteins, lipids and complex carbohydrates (polysaccharides), nucleic acids are one of the four major types of macromolecules that are essential for all known forms of life.
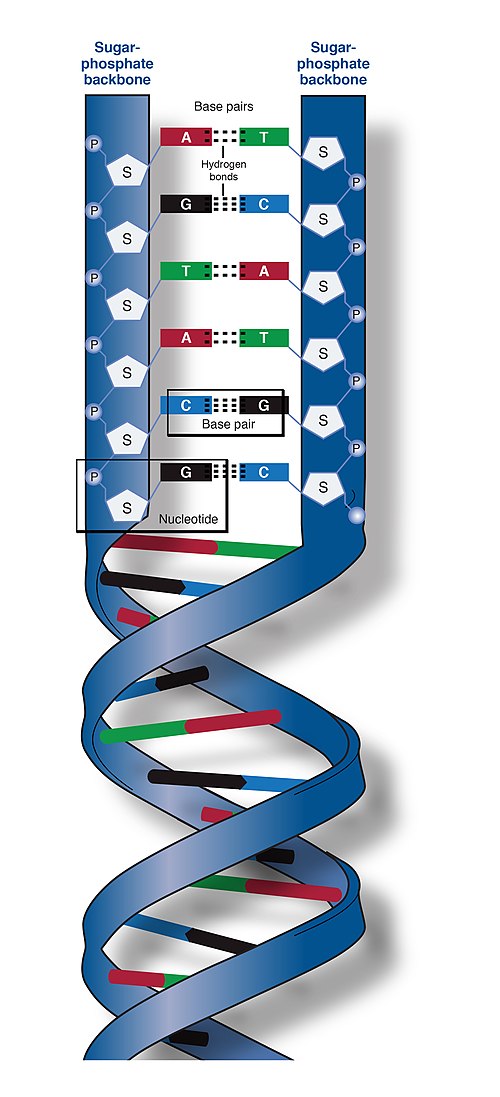
Simplified diagram

Impure DNA extracted from an orange

T7 RNA polymerase (blue) producing an mRNA (green) from a DNA template (orange)

A current model of meiotic recombination, initiated by a double-strand break or gap, followed by pairing with an homologous chromosome and strand invasion to initiate the recombinational repair process. Repair of the gap can lead to crossover (CO) or non-crossover (NCO) of the flanking regions. CO recombination is thought to occur by the Double Holliday Junction (DHJ) model, illustrated on the right, above. NCO recombinants are thought to occur primarily by the Synthesis Dependent Strand Annealing (SDSA) model, illustrated on the left, above. Most recombination events appear to be the SDSA type.